Fludarabine Prescribing Information
Package insert / product label
Generic name: fludarabine phosphate
Dosage form: injection, solution
Drug class: Antimetabolites
Medically reviewed by Drugs.com. Last updated on Sep 6, 2023.
HEALTH CARE PROVIDER LETTER

July 18, 2023
Teva Administrative Offices:
Teva Pharmaceuticals USA, Inc.
Morris Corporate Center
400 Interpace Parkway
Parsippany, NJ 07054
Phone: 1-888-838-2872
Subject: Temporary Importation of Fludarabine Phosphate Injection, USP 50 mg per 2 mL (25 mg/mL),
Mitigation to Address Supply Shortage
Dear Health Care Provider:
To alleviate a critical shortage of Fludarabine Phosphate Injection, USP 50 mg per 2 mL (25 mg/mL) single dose vials in the United States (U.S.) market, Teva has coordinated with the U.S. Food and Drug Administration (FDA) to make available a temporary supply of Fludarabin (Fludarabine Phosphate Injection) Actavis, single dose vials, that is not currently approved under Teva’s ANDA (203738). The lots for this temporary supply of Fludarabin Actavis 50 mg per 2 mL (25 mg/mL) marketed in Sweden are manufactured at the same Teva facility that supplied product for the U.S. market. These lots have undergone an internal review by Teva and met all quality specifications.
Effective immediately, Teva will distribute the following presentation of Fludarabin Actavis in the U.S. to address the critical shortage:
|
Product Name and Description |
Size |
Marketing Authorization Number |
NDC* |
Lot Number(s) |
|
Fludarabin (Fludarabine Phosphate Injection) Actavis, single dose vials |
50 mg/2 mL (25 mg/mL) |
48182 |
0480-9772-01 |
2NM5013 2NM5022 |
*NDC has been assigned for purposes of supply chain systems product identification and processing only. NDC does NOT appear on the product packaging itself, nor is it encoded in any barcoding.
Indication for Fludarabin Actavis
Fludarabine Phosphate Injection, USP is indicated for the treatment of adult patients with B-cell chronic lymphocytic leukemia (CLL) who have not responded to or whose disease has progressed during treatment with at least one standard alkylating-agent containing regimen. The safety and effectiveness of Fludarabine Phosphate Injection, USP in previously untreated or non-refractory patients with CLL have not been established.
Please refer to the FDA-approved Fludarabine Phosphate Injection prescribing information available on DailyMed at DailyMed - FLUDARABINE PHOSPHATE injection, solution (nih.gov) and follow the instructions presented in the FDA-approved package insert, except as noted below about diluting the product in saline (0.9% Sodium Chloride). After June 30, 2023, the U.S. Fludarabine Phosphate Injection prescribing information will be available in the DailyMed Labeling Archives at https://dailymed.nlm.nih.gov/dailymed/archives/index.cfm?query=a7743c62-2230-4289-92dc-6fc9c61f41d2&date=.
Important Information Regarding Fludarabin Actavis
There is one important difference in preparation between the FDA-approved product and the supplied product:
- When preparing the Fludarabin Actavis for intravenous administration, the product should only be diluted in saline (0.9% Sodium Chloride) as the stability of diluted EU product has only been tested in saline (0.9% Sodium Chloride).
However, there are no clinically relevant differences in the indication or safety information between the approved U.S package insert and that of the supplied product, or in how healthcare professionals will prescribe or dispense Fludarabin Actavis. Please see the product label comparison table at the end of this letter.
Fludarabin Actavis will be available only by prescription in the U.S. However, the imported lots do not have the statement “Rx only” on their labeling.
There is no barcode on this product for use with U.S. barcode scanning systems. Alternative procedures should be followed to ensure that the correct drug product is being used and administered to individual patients.
In addition, the packaging of the Fludarabin Actavis does not include serialization information and does not meet the product identifier requirements of section 582(b)(2) of the Federal Food, Drug and Cosmetic Act.
Reporting Adverse Events
Healthcare providers should report adverse events associated with the use of Fludarabin Actavis Injection to Teva at 1-888-838-2872, Option 3 and then Option 4.
Adverse events, medication errors or quality problems experienced with the use of this product may also be reported to the FDA’s MedWatch Adverse Event Reporting Program either online, by regular mail, or by fax:
- Complete and submit the report Online: www.fda.gov/medwatch/report.htm
- Regular mail or Fax: Download form www.fda.gov/MedWatch/getforms.htm or call 1-800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form or submit by fax to 1-800-FDA-0178 (1-800-332-0178).
If you have any questions about the information contained in this letter, any quality related problems, or questions on the use of Fludarabin Actavis 25 mg/mL single dose vials, please contact Teva Pharmaceuticals USA, Inc. at 1-888-838-2872.
To place an order, please contact Teva directly at 1-888-838-2872, select option 3, then select option 9.
We remain at your disposal to answer any questions you may have about our product and to provide more information if needed.
Sincerely,

Denisa Hurtukova, MD
Vice President, Head of North America Medical Affairs
Teva Pharmaceuticals
Related/similar drugs
Imbruvica, Jaypirca, Venclexta, prednisone, methotrexate, triamcinolone, dexamethasone
Product Label and Product Characteristics Side-by-Side Comparison Table
| Carton (US FDA Approved) | |
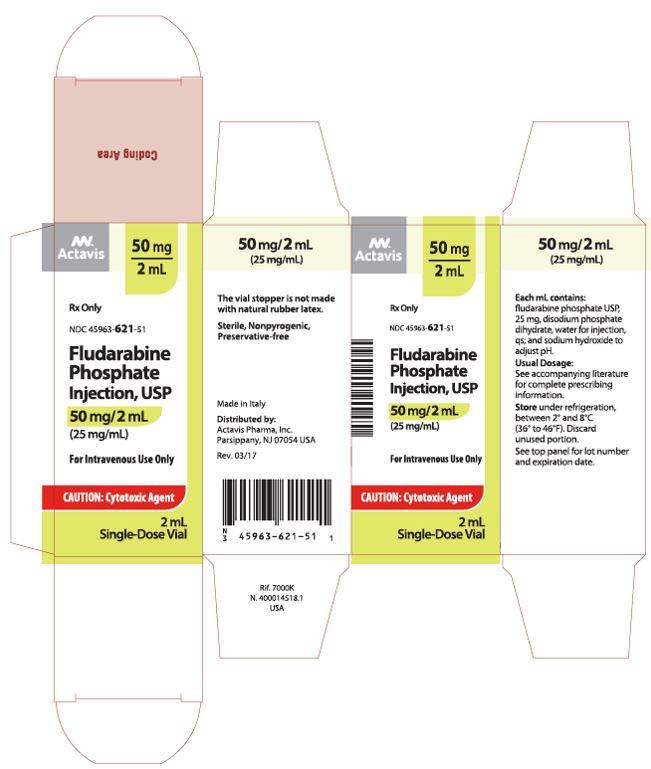 |
|
| Carton (Import Product) | |
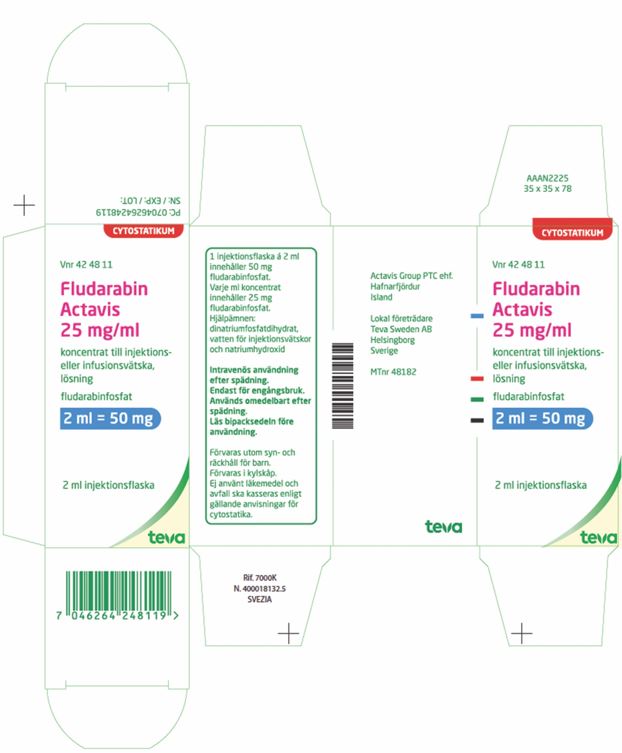 |
|
|
Vial Label |  |
|
Vial Label (Import Product) |  |
|
PRESCRIBING INFORMATION |
||
|
US FDA Approved Product |
Import Product |
|
|
INDICATION |
Fludarabine Phosphate Injection, USP is a nucleotide metabolic inhibitor indicated for: |
4.1 Therapeutic indications |
|
DOSAGE AND ADMINISTRATION |
2.1 Recommended Dose
Creatinine Clearance Starting: Dose |
4.2 Posology and method of administration Posology Doses should be adjusted for patients with reduced kidney function. If creatinine clearance is between 30 and 70 ml/min, the dose should be reduced by up to 50% and close haematological monitoring should be used to assess toxicity (see section 4.4).
|
| DESCRIPTION |
11 DESCRIPTION Figure 1: Chemical Structure of Fludarabine Phosphate 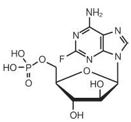
| 2. QUALITATIVE AND QUANTITATIVE COMPOSITION Each ml of concentrate for solution for injection or infusion contains 25 mg fludarabine phosphate. Each vial of 2 ml contains 50 mg fludarabine phosphate. For the full list of excipients, see section 6.1. 3. PHARMACEUTICAL FORM Concentrate for solution for injection or infusion.
6.1 List of excipients This medicinal product must not be mixed with other medicinal products except those mentioned in section 6.6. |
| HOW SUPPLIED/STORAGE AND HANDLING |
16.1 How Supplied Store under refrigeration, between 2° and 8°C (36° to 46°F). Procedures for proper handling and disposal should be considered. Consideration should be given to handling and disposal according to guidelines issued for cytotoxic drugs. Several guidelines on this subject have been published. 1-4 Caution should be exercised in the handling and preparation of Fludarabine Phosphate Injection solution. The use of latex gloves and safety glasses is recommended to avoid exposure in case of breakage of the vial or other accidental spillage. If the solution contacts the skin or mucous membranes, wash thoroughly with soap and water; rinse eyes thoroughly with plain water. Avoid exposure by inhalation or by direct contact of the skin or mucous membranes. The vial stopper is not made with natural rubber latex. | 6.3 Shelf life Vial before opening: 3 years After dilution: The diluted solution of /.../ in 0.9% sodium chloride is stable for up to 28 days in PVC and PE bags at Store between 2-8°C. Colourless glass vial (type I) with bromobutylic rubber stopper and metallic cap (aluminium) with polypropylene disk. Vial will be packed with or without a protective plastic overwrap. 2 ml vial
Dilution Only clear, colourless to yellowish solutions without particles should be used. /.../ should not be used in case of a defective container. /.../ should not be handled by pregnant staff. Procedures for proper handling should be followed according to local requirements for cytotoxic drugs. Caution should be exercised in the handling and preparation of the /.../ solution. The use of protective gloves and safety glasses is recommended to avoid exposure in case of breakage of the vial or other accidental spillage. |
| CONTRAINDICATIONS | None |
4.3 Contraindications
|
|
WARNINGS AND |
Boxed Warning
WARNING: SEVERE BONE MARROW SUPPRESSION, CNS TOXICITY, HEMOLYTIC ANEMIA, AND PULMONARY TOXICITY
Fludarabine Phosphate Injection should be administered under the supervision of a qualified physician experienced in the use of antineoplastic therapy. Fludarabine phosphate injection can severely suppress bone marrow function. When used at high doses in dose-ranging studies in patients with acute leukemia, fludarabine phosphate injection was associated with severe neurologic effects, including blindness, coma, and death. This severe central nervous system toxicity occurred in 36% of patients treated with doses approximately four times greater (96 mg/m2/day for 5 to 7 days) than the recommended dose. Similar severe central nervous system toxicity, including coma, seizures, agitation and confusion, has been reported in patients treated at doses in the range of the dose recommended for chronic lymphocytic leukemia [see Warnings and Precautions (5.2)].
Instances of life-threatening and sometimes fatal autoimmune phenomena such as hemolytic anemia, autoimmune thrombocytopenia/thrombocytopenic purpura (ITP), Evans syndrome, and acquired hemophilia have been reported to occur after one or more cycles of treatment with fludarabine phosphate injection. Patients undergoing treatment with Fludarabine Phosphate Injection should be evaluated and closely monitored for hemolysis [see Warnings and Precautions (5.3)].
In a clinical investigation using fludarabine phosphate in combination with pentostatin (deoxycoformycin) for the treatment of refractory chronic lymphocytic leukemia (CLL), there was an unacceptably high incidence of fatal pulmonary toxicity. Therefore, the use of Fludarabine Phosphate Injection in combination with pentostatin is not recommended [see Warnings and Precautions (5.5)].
There are clear dose dependent toxic effects seen with fludarabine phosphate. Dose levels approximately 4 times greater (96 mg/m2/day for 5 to 7 days) than that recommended for CLL (25 mg/m2/day for 5 days) were associated with a syndrome characterized by delayed blindness, coma and death. Symptoms appeared from 21 to 60 days following the last dose. Thirteen of 36 patients (36%) who received fludarabine phosphate at high doses
Severe bone marrow suppression, notably anemia, thrombocytopenia and neutropenia, has been reported in patients treated with fludarabine phosphate. In a Phase I study in adult solid tumor patients, the median time to nadir counts was 13 days (range, 3 to 25 days) for granulocytes and 16 days (range, 2 to 32 days) for platelets. Most patients had hematologic impairment at baseline either as a result of disease or as a result of prior myelosuppressive therapy. Cumulative myelosuppression may be seen. While chemotherapy-induced myelosuppression is often reversible, administration of Fludarabine Phosphate Injection requires careful hematologic monitoring.
Instances of life-threatening and sometimes fatal autoimmune phenomena such as hemolytic anemia, autoimmune thrombocytopenia/thrombocytopenic purura (ITP), Evans syndrome, and acquired hemophilia have been reported to occur after one or more cycles of treatment with fludarabine phosphate in patients with or without a previous history of autoimmune hemolytic anemia or a positive Coombs' test and who may or may not be in remission from their disease. Steroids may or may not be effective in controlling these hemolytic episodes. The majority of patients rechallenged with fludarabine phosphate developed a recurrence in the hemolytic process. The mechanism(s) which predispose patients to the development of this complication has not been identified. Patients undergoing treatment with Fludarabine Phosphate Injection should be evaluated and closely monitored for hemolysis. Discontinuation of therapy with Fludarabine Phosphate Injection is recommended in case of hemolysis. Transfusion-associated graft-versus-host disease has been observed after transfusion of non-irradiated blood in fludarabine phosphate treated patients. Fatal outcome as a consequence of this disease has been reported. Therefore, to minimize the risk of transfusion-associated graft-versus-host disease, patients who require blood transfusion and who are undergoing, or who have received, treatment with Fludarabine Phosphate Injection should receive irradiated blood only. In a clinical investigation using fludarabine phosphate in combination with pentostatin (deoxycoformycin) for the treatment of refractory chronic lymphocytic leukemia (CLL) in adults, there was an unacceptably high incidence of fatal pulmonary toxicity. Therefore, the use of Fludarabine Phosphate Injection in combination with pentostatin is not recommended.
Pregnancy Category D Males with female sexual partners of childbearing potential should use contraception during and after cessation of fludarabine phosphate therapy. Fludarabine phosphate may damage testicular tissue and spermatozoa. Possible sperm DNA damage raises concerns about loss of fertility and genetic abnormalities in fetuses. The duration of this effect is uncertain [see Nonclinical Toxicology (13.1)]. Tumor lysis syndrome has been associated with fludarabine phosphate treatment. This syndrome has been reported in CLL patients with large tumor burdens. Since fludarabine phosphate can induce a response as early as the first week of treatment, precautions should be taken in those patients at risk of developing this complication. Fludarabine Phosphate Injection must be administered cautiously in patients with renal impairment. The total body clearance of 2-fluoro-ara-A has been shown to be directly correlated with creatinine clearance. Patients with creatinine clearance 30 to 79 mL/min should have their fludarabine phosphate dose reduced and be monitored closely for excessive toxicity. Fludarabine phosphate should not be administered to patients with creatinine clearance less than 30 mL/min [see Dosage and Administration (2.2) and Use in Specific Populations (8.6)].
During and after treatment with Fludarabine Phosphate Injection, vaccination with live vaccines should be avoided. | No Boxed Warning 4.4 Special warnings and precautions for use
Myelosuppression Irrespective of any previous history of autoimmune processes or Coombs test status, life-threatening and sometimes fatal autoimmune phenomena (see section 4.8) have been reported to occur during or after treatment with fludarabine phosphate. The majority of patients experiencing haemolytic anaemia developed a recurrence in the haemolytic process after rechallenge with fludarabine phosphate. Patients treated with fludarabine phosphate should be closely monitored for signs of haemolysis. The effect of chronic administration of fludarabine on the central nervous system is unknown. However, patients tolerated the recommended dose in some studies for relatively long treatment times (for up to 26 courses of therapy).
LE, ATL or RPLS symptoms may include headache, nausea and vomiting, seizures, visual disturbances such as vision loss, altered sensorium, and focal neurological deficits. Additional effects may include optic neuritis, and papillitis, confusion, somnolence, agitation, paraparesis/ quadriparesis, muscle spasticity and incontinence. Tumour lysis syndrome has been reported in CLL patients with large tumour burdens. Since fludarabine phosphate can induce a response as early as the first week of treatment, precautions should be taken in those patients at risk of developing this complication, and hospitalisation may be recommended for these patients during the first course of treatment. The worsening or flare up of pre-existing skin cancer lesions as well as new onset of skin cancer have been reported in some patients during or after fludarabine phosphate therapy. In patients with impaired state of health, fludarabine phosphate should be given with caution and after careful risk/benefit consideration. This applies especially for patients with severe impairment of bone marrow function (thrombocytopenia, anaemia, and/or granulocytopenia), immunodeficiency or with a history of opportunistic infection. The total body clearance of the principle plasma metabolite 2-F-ara-A shows a correlation with creatinine clearance, indicating the importance of the renal excretion pathway for the elimination of the compound. Patients with reduced renal function demonstrated an increased total body exposure (AUC of 2F-ara-A). There are limited clinical data available in patients with impairment of renal function (creatinine clearance <70 ml/min). In patients with hepatic impairment fludarabine phosphate should be used with caution because it can cause hepatic toxicity. Fludarabine phosphate should only be administered if the perceived benefit outweighs any potential risk. Such patients should be monitored closely for excessive toxicity and dosage should be modified or the treatment discontinued accordingly. See also section 4.2. Since there are limited data for the use of fludarabine phosphate in elderly persons >75 years), caution should be exercised with the administration of fludarabine phosphate in these patients (see also section 4.2). Fludarabine phosphate should not be used during pregnancy unless clearly necessary (e.g. lifethreatening situation, no alternative safer treatment available without compromising the therapeutic benefit, treatment cannot be avoided). It has the potential to cause foetal harm (see sections 4.6 and 5.3). Prescribers may only consider the use of fludarabine, if the potential benefits justify the potential risks to the foetus. During and after treatment with fludarabine phosphate vaccination with live vaccines should be avoided. A crossover from initial treatment with fludarabine phosphate to chlorambucil for non-responders to fludarabine phosphate should be avoided because most patients who have been resistant to fludarabine phosphate have shown resistance to chlorambucil.
Excipients Fludarabine phosphate may reduce the ability to drive and use machines since e.g. fatigue, weakness, visual disturbances, confusion, agitation and seizures have been observed. |
| ADVERSE REACTIONS |
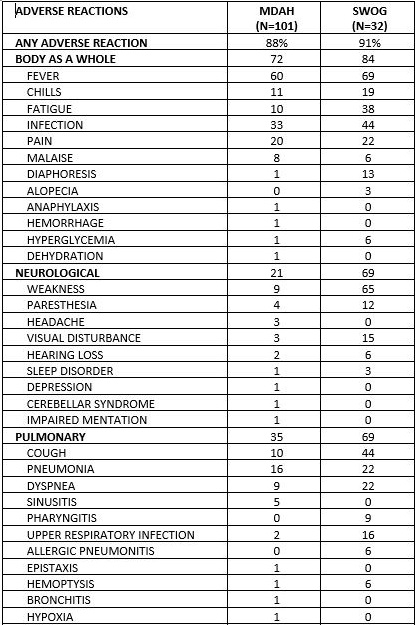
6 ADVERSE REACTIONS Hematologic events (neutropenia, thrombocytopenia, and/or anemia) were reported in the majority of CLL patients treated with fludarabine phosphate. During fludarabine phosphate treatment of 133 patients with CLL, the absolute neutrophil count decreased to less than 500/mm3 in 59% of patients, hemoglobin decreased from pretreatment values by at least 2 grams percent in 60%, and platelet count decreased from pretreatment values by at least 50% in 55%. Myelosuppression may be severe, cumulative, and may affect multiple cell lines. Bone marrow fibrosis occurred in one CLL patient treated with fludarabine phosphate.
Serious and sometimes fatal infections, including opportunistic infections and reactivations of latent viral infections such as VZV (herpes zoster), Epstein-Barr virus and JC virus (progressive multifocal leukoencephalopathy) have been reported in patients treated with fludarabine phosphate.
Tumor lysis syndrome has been reported in CLL patients treated with fludarabine phosphate. This complication may include hyperuricemia, hyperphosphatemia, hypocalcemia, metabolic acidosis, hyperkalemia, hematuria, urate crystalluria, and renal failure. The onset of this syndrome may be heralded by flank pain and hematuria. Objective weakness, agitation, confusion, seizures, visual disturbances, optic neuritis, optic neuropathy, blindness and coma have occurred in CLL patients treated with fludarabine phosphate at the recommended dose. Peripheral neuropathy has been observed in patients treated with fludarabine phosphate and one case of wrist-drop was reported. There have been additional reports of cerebral hemorrhage though the frequency is not known [see Warnings and Precautions (5)]. Pneumonia, a frequent manifestation of infection in CLL patients, occurred in 16%, and 22% of those treated with fludarabine phosphate in the MDAH and SWOG studies, respectively. Pulmonary hypersensitivity reactions to fludarabine phosphate characterized by dyspnea, cough and interstitial pulmonary infiltrate have been observed.
Gastrointestinal disturbances such as nausea, vomiting, anorexia, diarrhea, stomatitis, and hemorrhage have been reported in patients treated with fludarabine phosphate. Elevations of pancreatic enzyme levels have also been reported.
Hemorrhagic cystitis has been reported in patients treated with fludarabine phosphate. Skin toxicity, consisting primarily of skin rashes, has been reported in patients treated with fludarabine phosphate. Erythema multiforme, Steven-Johnson syndrome, toxic epidermal necrolysis and pemphigus have been reported, with fatal outcomes in some cases. Worsening or flare-up of pre-existing skin cancer lesions, as well as new onset of skin cancer, has been reported in patients during or after treatment with fludarabine phosphate. Elevations of hepatic enzyme levels have been reported. Data in Table 1 are derived from the 133 patients with CLL who received fludarabine phosphate in the MDAH and SWOG studies. Table 1: PERCENT OF CLL PATIENTS REPORTING NON-HEMATOLOGIC ADVERSE REACTIONS
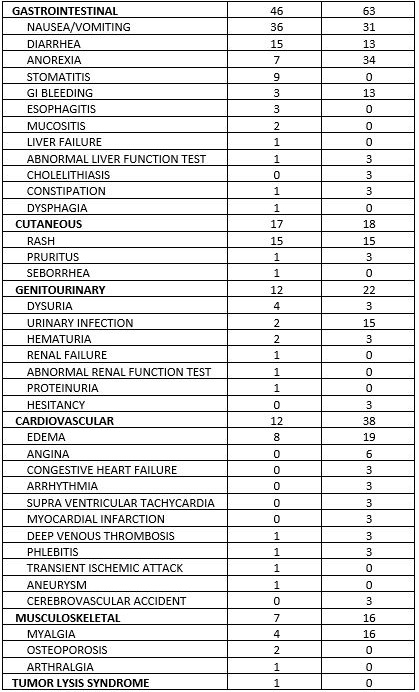
| 4.8 Undesirable effects Summary of safety profile Based on the experience with the use of fludarabine phosphate, the most common adverse events include myelosuppression (neutropenia, thrombocytopenia and anaemia), infection including pneumonia, cough, fever, fatigue, weakness, nausea, vomiting and diarrhoea. Other commonly reported events include chills, oedema, malaise, peripheral neuropathy, visual disturbance, anorexia, mucositis, stomatitis and skin rash. Serious opportunistic infections have occurred in patients treated with fludarabine phosphate. Fatalities as a consequence of serious adverse events have been reported. Tabulated list of adverse reactions The table below reports adverse events by MedDRA system organ classes (MedDRA SOCs). The frequencies are based on clinical trial data regardless of the causal relationship with fludarabine. The rare adverse reactions were mainly identified from the post-marketing experience. 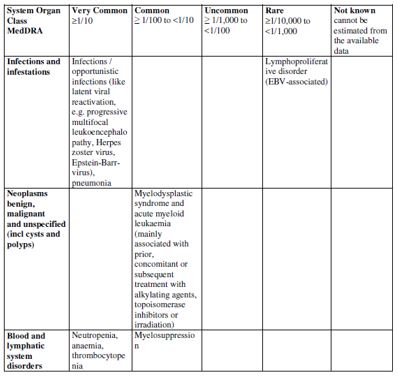 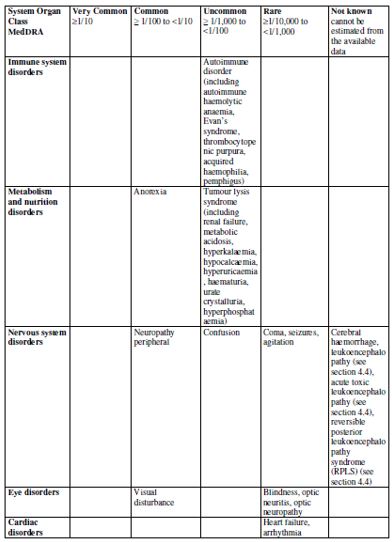 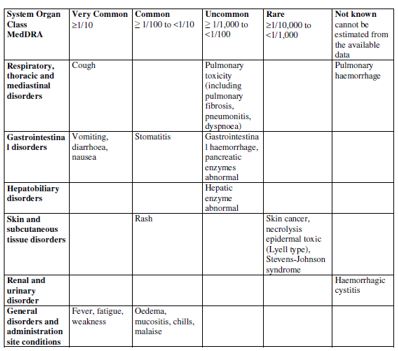 The most appropriate MedDRA term to describe a certain adverse event is listed. Synonyms or related conditions are not listed, but should be taken into account as well. Adverse event term representation is based on MedDRA version 12.0.
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system listed in Appendix V. |
| 7 DRUG INTERACTIONS | 7.1 Pentostatin | 4.5 Interaction with other medicinal products and other forms of interaction
In a clinical investigation using intravenous fludarabine phosphate in combination with pentostatin (deoxycoformycin) for the treatment of refractory chronic lymphocytic leukaemia (CLL), there was an unacceptably high incidence of fatal pulmonary toxicity. Therefore, the use of fludarabine phosphate in combination with pentostatin is not recommended. Dipyridamole and other inhibitors of adenosine uptake may reduce the therapeutic efficacy of fludarabine phosphate. Clinical studies and in vitro experiments showed that during use of fludarabine in combination with cytarabine the intracellular peak concentration and intracellular exposure of Ara-CTP (active metabolite of cytarabine) increased in leukemic cells. Plasma concentrations of Ara-C and the elimination rate of Ara-CTP were not affected. |
| 8 USE IN SPECIFIC POPULATIONS |
8.1 Pregnancy
It is not known whether fludarabine phosphate is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions including tumorigenicity in nursing infants, a decision should be made to discontinue nursing or discontinue the drug, taking into account the importance of the drug for the mother. Data submitted to the FDA was insufficient to establish efficacy in any childhood malignancy.
The total body clearance of the principal metabolite 2-fluoro-ara-A correlated with the creatinine clearance, indicating the importance of the renal excretion pathway for the elimination of the drug. Renal clearance represents approximately 40% of the total body clearance. Patients with creatinine clearance 30 to 79 mL/min should have their fludarabine phosphate dose reduced and be monitored closely for excessive toxicity. Due to insufficient data, fludarabine phosphate should not be administered to patients with creatinine clearance less than 30 mL/min [see Dosage and Administration |
4.6 Fertility, pregnancy and lactation Preclinical data in rats demonstrated a transfer of fludarabine and/or metabolites through the placenta. The results from intravenous embryotoxicity studies in rats and rabbits indicated an embryolethal and teratogenic potential at the therapeutic doses (see section 5.3). It is not known whether this drug or its metabolites are excreted in human milk. |
| 10 OVERDOSAGE | High doses of fludarabine phosphate [see Warnings and Precautions (5)] have been associated with an irreversible central nervous system toxicity characterized by delayed blindness, coma and death. High doses are also associated with severe thrombocytopenia and neutropenia due to bone marrow suppression. There is no known specific antidote for fludarabine phosphate overdosage. Treatment consists of drug discontinuation and supportive therapy. | 4.9 Overdose
High doses of fludarabine phosphate have been associated with leukoencephalopathy, acute toxic leukoencephalopathy, or reversible posterior leukoencephalopathy syndrome (RPLS). Symptoms may include headache, nausea and vomiting, seizures, visual disturbances such as vision loss, altered sensorium, and focal neurological deficits. Additional effects may include optic neuritis, and papillitis, confusion, somnolence, agitation, paraparesis/ quadriparesis, muscle spasticity, incontinence, irreversible central nervous system toxicity characterised by delayed blindness, coma, and death. High doses are also associated with severe thrombocytopenia and neutropenia due to bone marrow suppression. |
| CLINICAL PHARMACOLOGY |
12.1 Mechanism of Action Phase I studies in humans have demonstrated that fludarabine phosphate is rapidly converted to the active metabolite, 2-fluoro-ara-A, within minutes after intravenous infusion. Consequently, clinical pharmacology studies have focused on 2-fluoro-ara-A pharmacokinetics. After the five daily doses of 25 mg 2-fluoro-ara-AMP/m2 to cancer patients infused over 30 minutes, 2-fluoro-ara-A concentrations show a moderate accumulation. During a 5-day treatment schedule, 2-fluoroara-A plasma trough levels increased by a factor of about 2. The terminal half-life of 2-fluoro-ara-A was estimated as approximately 20 hours. In vitro, plasma protein binding of fludarabine ranged between 19% and 29%. A correlation was noted between the degree of absolute granulocyte count nadir and increased area under the concentration x time curve (AUC). |
5. PHARMACOLOGICAL PROPERTIES The pharmacokinetics of fludarabine (2F-ara-A) have been studied after intravenous administration by rapid bolus injection and short-term infusion as well as following continuous infusion of fludarabine phosphate (fludarabine phosphate, 2F-ara-AMP). 2F-ara-AMP is a water-soluble prodrug of fludarabine (2F-ara-A), which is rapidly and quantitatively dephosphorylated in the human organism to the nucleoside fludarabine (2F ara-A). 2F-ara-A elimination is largely by renal excretion. 40 to 60 % of the administered i.v. dose was excreted in the urine. Mass balance studies in laboratory animals with ³H-2F-ara-AMP showed a complete recovery of radio-labelled substances in the urine. Individuals with impaired renal function exhibited a reduced total body clearance, indicating the need for a dose reduction. In vitro investigations with human plasma proteins revealed no pronounced tendency of 2F-ara-A protein binding. 2F-ara-A is actively transported into leukaemic cells, whereupon it is rephosphorylated to the |
|
13 NONCLINICAL |
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility | 5.3 Preclinical safely data Systemic toxicity In acute toxicity studies, single doses of fludarabine phosphate produced severe intoxication symptoms or death at dosages about two orders of magnitude above the therapeutic dose. As expected for a cytotoxic compound, the bone marrow, lymphoid organs, gastrointestinal mucosa, kidneys and male gonads were affected. In patients, severe side effects were observed closer to the recommended therapeutic dose (factor 3 to 4) and included severe neurotoxicity partly with lethal outcome (see section 4.9).
|
| 14 CLINICAL STUDIES |
14.1 Adults
| Clinical efficacy and safety A phase III trial in patients with previously untreated B-chronic lymphocytic leukaemia comparing treatment with fludarabine phosphate vs. chlorambucil (40 mg / m² q4 weeks) in 195 and 199 patients respectively showed the following outcome: statistically significant higher overall response rates and complete response rates after 1st line treatment with fludarabine phosphate compared to chlorambucil (61.1% vs. 37.6% and 14.9% vs. 3.4%, respectively); statistically significant longer duration of response (19 vs. 12.2 months) and time to progression (17 vs. 13.2 months) for the patients in the fludarabine phosphate group. The median survival of the two patient groups was 56.1 months for fludarabine phosphate and 55.1 months for chlorambucil, a non-significant difference was also shown with performance status. The proportion of patients reported to have toxicities were comparable between fludarabine phosphate patients (89.7%) and chlorambucil patients (89.9%). While the difference in the overall incidence of haematological toxicities was not significant between the two treatment groups, significantly greater proportions of fludarabine phosphate patients experienced white blood cell (p=0.0054) and lymphocyte (p=0.0240) toxicities than chlorambucil patients. The proportions of patients who experienced nausea, vomiting, and diarrhoea were significantly lower for fludarabine phosphate patients (p<0.0001, p<0.0001, and p=0.0489, respectively) than chlorambucil patients. Toxicities of the liver were also reported for significantly (p=0.0487) less proportions of patients in the fludarabine phosphate group than in the chlorambucil group. Patients who initially respond to fludarabine phosphate have a chance of responding again to fludarabine phosphate monotherapy.
|
| 17 PATIENT COUNSELING INFORMATION |
17.1 Monitoring During treatment, the patient's hematologic profile (particularly neutrophils, red blood cells, and platelets) should be monitored regularly to determine the degree of hematopoietic suppression [see Warnings and Precautions (5.2)]. | |
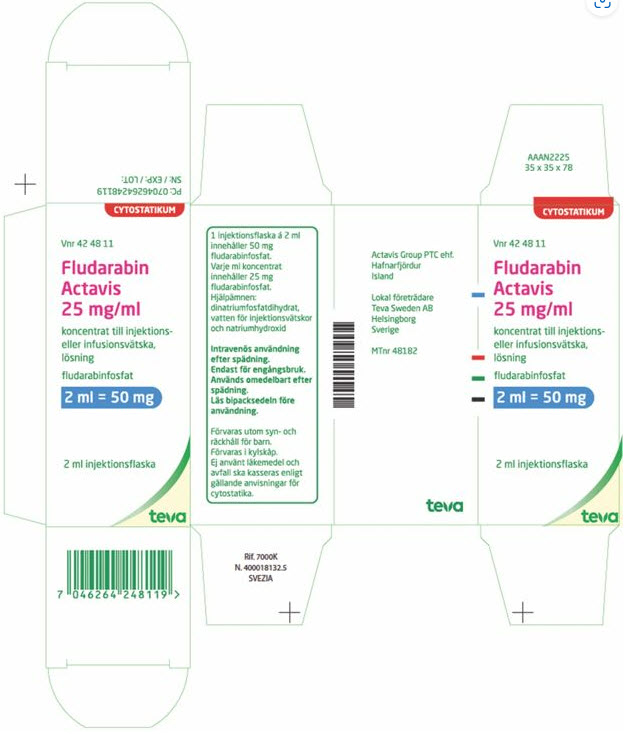
PRINCIPAL DISPLAY PANEL
Vnr 42 48 11
CYTOSTATIKUM
Fludarabin
Actavis
25 mg/ml
koncetrat till injektions-
eller infusionsvätska,
lösning
fludarabinfosfat
2 ml = 50 mg
2 ml injektionsflaska
English Translation
CYTOSTATICS
No. 42 48 11
Fludarabin
Actavis
25 mg/ml
concentrate for solution for injection or infusion
fludarabin phosphate
2 ml = 50 mg
2 ml vial
| FLUDARABINE PHOSPHATE
fludarabine phosphate injection, solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Teva Pharmaceuticals, Inc. (022629579) |
More about fludarabine
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (1)
- Side effects
- Dosage information
- During pregnancy
- Drug class: antimetabolites
- En español
Patient resources
- Fludarabine injection drug information
- Fludarabine (Intravenous) (Advanced Reading)
- Fludarabine (Oral) (Advanced Reading)
